
Le craniosinostosi
Le craniosinostosi sono responsabili di un gruppo eterogeneo di disordini dello sviluppo caratterizzato da anomalie nella forma del cranio; la loro comparsa è indotta da una prematura chiusura di una o più suture craniche.
La configurazione del cranio nelle craniosinostosi è il risultato della distribuzione anatomica delle suture e dall’ordine con il quale queste suture si chiudono; la loro prematura chiusura porta alla crescita compensatoria delle regioni non colpite e quindi ad anormalità facciali e della calotta cranica. Questo processo di ossificazione prematura, soprattutto nei casi più severi, produce frequentemente ernie intracraniche e può compromettere le normali funzioni respiratorie, visive, uditive e intellettive.
Complessivamente le craniosinostosi si manifestano con una incidenza relativamente alta, stimata essere circa 1 su 2500 nati.
Sono stati riscontrati sia casi sporadici che familiari che delineano oltre 100 differenti forme di craniosinostosi che possono manifestarsi come condizione isolata o associata ad un caratteristico quadro sindromico.
In più della metà di queste condizioni craniosinostotiche è stata ipotizzata e in alcuni casi stabilita, una base genetica, per lo più di tipo monogenica a trasmissione autosomica dominante.
La comprensione delle basi genetiche di questi disordini dello sviluppo sta cominciando ad essere delineata da recenti studi in cui si è dimostrato che alcuni recettori di membrana, quali quelli per i fattori di crescita dei fibroblasti (FGFR) e fattori di trascrizione, quali MSX2 e TWIST, giocano un ruolo significativo nello sviluppo degli arti e in quello craniofacciale e che alcune delle sindromi craniosinostotiche sono associate a mutazioni in questi geni.
Il processo di sviluppo dello scheletro craniofacciale inizia dalla migrazione delle cellule della cresta neurale che si spostano dai rombomeri del cervello posteriore verso gli archi branchiali.
Queste cellule migrate nei processi craniofacciali danno origine ad un mesenchima addensato, che attraverso i processi di ossificazione endocondrale e intramembranosa, si differenzierà rispettivamente in cartilagine e osso del cranio.
La regolazione del disegno e del mantenimento della forma dello scheletro avviene in due tempi (Erlebacher et al., 1995). L’espressione di geni omeotici (Hox) in una prima fase trasmette alle cellule delle linee mesenchimali l’informazione di specificare l’intero disegno dello scheletro, la forma e l’identità di ogni elemento. In esperimenti di inattivazione di specifici geni omeotici è stato dimostrato il coinvolgimento di questi geni nelle prime fasi della specificazione della struttura dello scheletro craniofacciale.
In un secondo momento, tramite una regolazione locale della condensazione mesenchimale e la secrezione di fattori paracrini (molecole solubili che inducono modificazioni in cellule prossime spazialmente alla cellula secernente) e autocrini (molecole che producono cambiamenti nella cellula che li secerne), si definisce la forma degli elementi dello scheletro.
Mentre i geni Hox contribuiscono al “primo disegno” dello scheletro, i prodotti di un’altra famiglia di geni, le proteine della morfogenesi ossea (bone morfogenetic proteins, BMP) sembrano essere coinvolte nella regolazione della forma di ciascun elemento.
Fattori paracrini espressi nella prima fase della condensazione mesenchimale, le BMP influenzano l’ampiezza e la forma degli elementi dello scheletro (Mundlos et al., 1997). Questi fattori appartengono ad una grande famiglia di proteine ( la super famiglia del TGF-b fattore di crescita trasformante) in cui diversi membri risultano essere espressi a livello del fronte di ossificazione durante lo sviluppo delle suture craniche (Cohen, 1997).
Recentemente, i fattori di crescita dei fibroblasti (FGF) sono stati implicati nella coordinazione della crescita delle ossa. L’induzione del segnale dai recettori per gli FGF regola una moltitudine di processi cellulari, tra cui di maggior rilievo, la proliferazione e il differenziamento delle cellule di origine mesenchimale e neuroectodermica. Inoltre sia alterazioni nella struttura craniofacciale che anomalie a carico degli arti come nell’acondroplasia, la forma più comune di nanismo, sono state correlate a mutazioni nei geni per i recettori per gli FGF.
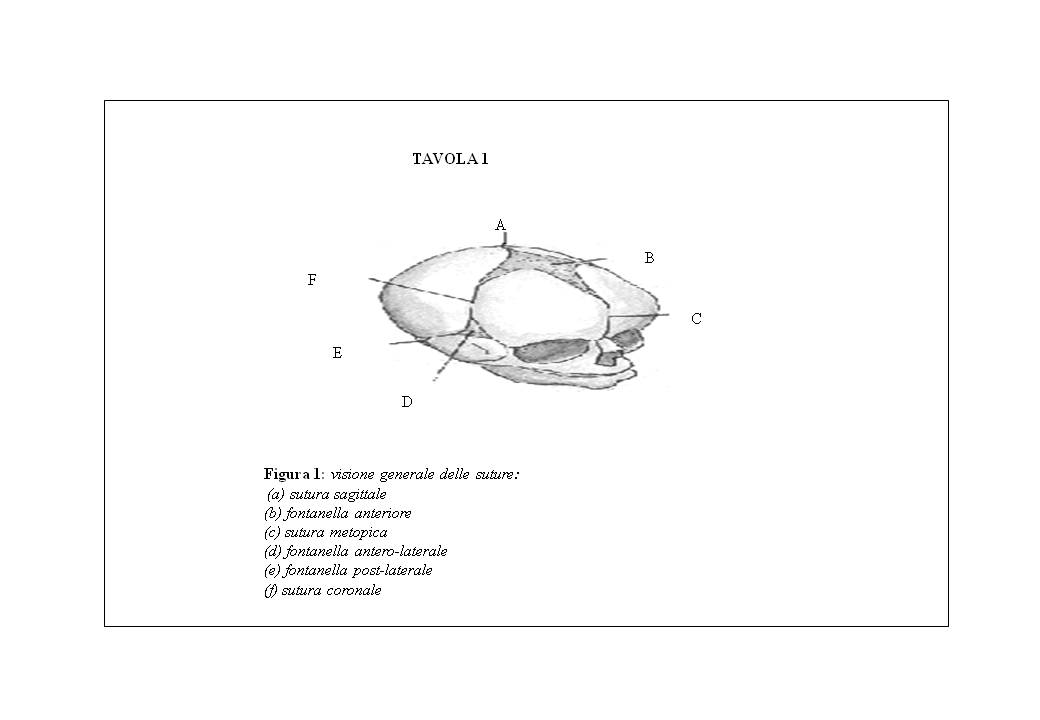
Esistono diverse definizioni del termine “sutura”. Nel Gray’s Anatomy (Goss,1959) è definita come “una articolazione nella quale margini contigui di ossa adiacenti sono uniti da un sottile strato di tessuto fibroso”. In Moss-Salentijn e Hendricks-Klyvert (1985) il concetto di sutura è descritto come “densa connessione fibrosa tra ossa adiacenti formate per via intramembranosa che permette il minor movimento”.
Le suture svolgono numerose funzioni, permettendo infatti la crescita ossea, provvedendo ad un collegamento stabile tra le ossa di nuova formazione e allo stesso tempo, permettendo alcuni movimenti fra di esse, consentono l’adattamento del cervello in espansione.
Le suture vengono distinte in (vedi tavola 1):
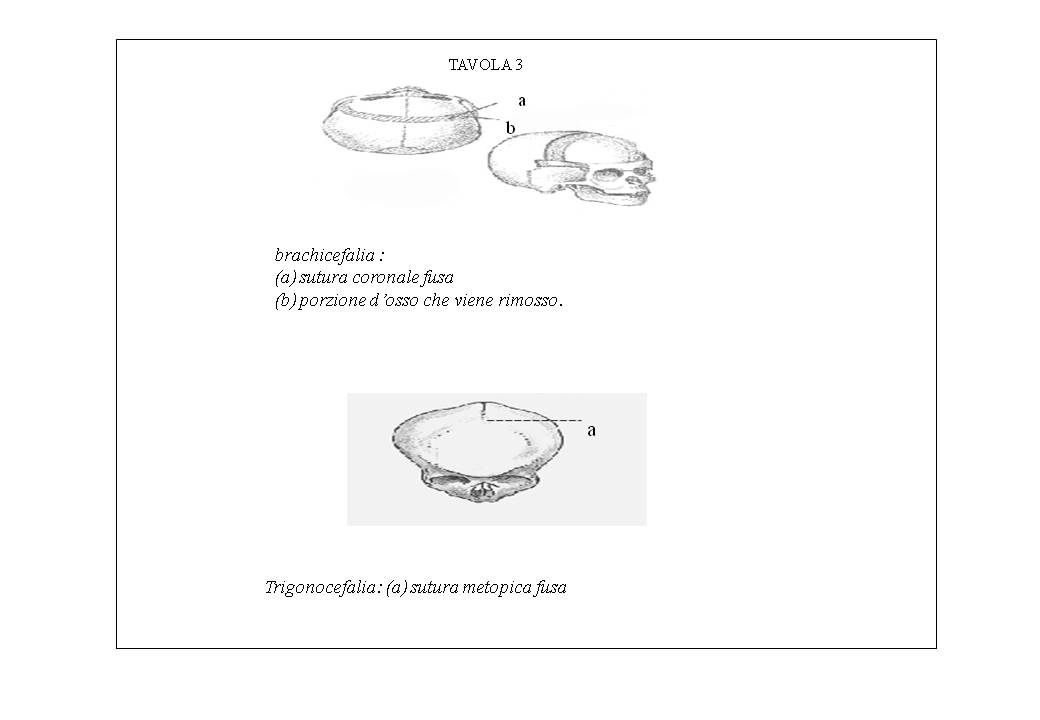
Scafocefalia (interparietale): è dovuta alla chiusura precoce della sutura sagittale ed è caratterizzata da cranio lungo e stretto a forma di barca, con prominenza delle bozze frontali. (tavola 2).
Pachicefalia (parieto-occipitale): è dovuta alla precoce chiusura delle suture lambdoidi, a volte associata con sinostosi multiple in particolare delle suture coronale e sagittale. La sua incidenza è molto bassa.
Plagiocefalia (sfeno-frontoparietale): è dovuta alla chiusura monolaterale della sutura coronale; è caratterizzata da cranio asimmetrico con appiattimento unilaterale della bozza frontale (tav 2).
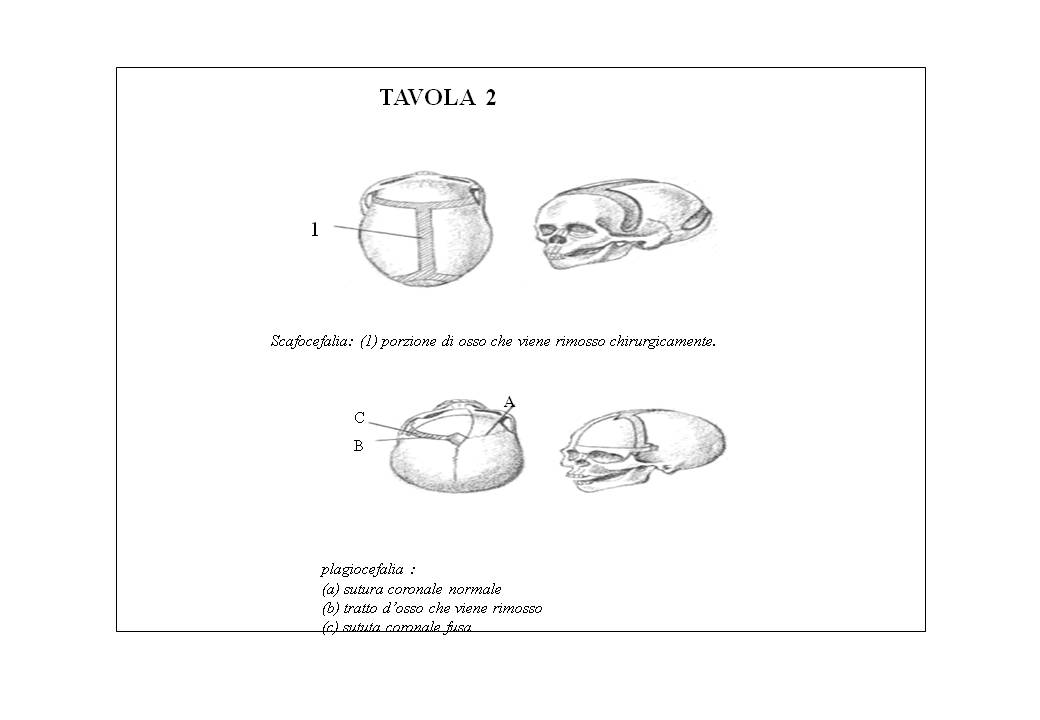
Brachicefalia (frontoparietale): è dovuta alla chiusura bilaterale della sutura coronale; è caratterizzata da cranio più corto nel piano sagittale e da una crescita compensatoria laterale nel senso della larghezza (tavola 3).
Trigonocefalia (interfrontale): è dovuta alla chiusura della sutura metopica; è caratterizzata da cranio a tre lobi con fronte stretta, protuberanza mediana, occhi ravvicinati (tavola 3). Sono stati riscontrati anche casi con ritardo mentale, strabismo, palato arcuato, polidattilismo, epicanto.
Turricefalia o Oxicefalia (frontointerparietale): è dovuta alla sinostosi delle ossa parietali con le ossa occipitali e temporali ed alla crescita compensatoria delle regioni anteriori delle fontanelle.
craniosinistosi
La sindrome di Crouzon è caratterizzata da una craniostenosi coronale e sagittale (acrobrachicefalia e oxicefalia), da una prominenza dell’osso frontale, e da una ipoplasia medio-facciale. I pazienti affetti da questa sindrome mostrano una notevole prominenza della fronte, ipoplasia del mascellare superiore, ipertelorismo, e naso adunco. Frequentemente sono associati disturbi della visione e malposizioni dentarie; può manifestarsi anche un lieve ritardo nello sviluppo intellettivo (Smith et al., 1982; Introzzi, 1984; Stricker et al., 1990; Murdoch-Kinch et al., 1997).
Le acrocefalosindattilie costituiscono un gruppo di sindromi a trasmissione autosomica dominante caratterizzate da craniostenosi della sutura coronale e anomalie distali degli arti. Questo gruppo comprende 4 sindromi principali: Apert, Saethre-Chotzen, Pfeiffer e Jackson-Weiss.
Nella sindrome di Apert le alterazioni più evidenti a livello del cranio sono la crescita in altezza della volta, che risulta più alta che larga, e l’appiattimento dell’osso occipitale. Le suture coronali e sagittali visibili alla nascita sono destinate a chiudersi precocemente. I soggetti affetti mostrano inoltre ipoplasia medio-facciale e frequentemente ipertelorismo. A livello degli arti è presente sindattilia delle mani e dei piedi che interessa principalmente 2°, 3°e 4° dito; il 1° dito presenta più o meno malformazioni. Oltre alle sindattilie, sono evidenti varie lesioni ossee come la fusione delle falangi distali che inoltre appaiano alterate; le falangi del pollice sono fuse e formano un unico osso quadrangolare. Spesso troviamo la fusione delle falangi in un unico osso anche nel 1° dito del piede, mentre le falangi delle altre dita mostrano le stesse anomalie di quelle della mano.
Nella sindrome di Apert le alterazioni più evidenti a livello del cranio sono la crescita in altezza della volta, che risulta più alta che larga, e l’appiattimento dell’osso occipitale. Le suture coronali e sagittali visibili alla nascita sono destinate a chiudersi precocemente. I soggetti affetti mostrano inoltre ipoplasia medio-facciale e frequentemente ipertelorismo. A livello degli arti è presente sindattilia delle mani e dei piedi che interessa principalmente 2°, 3°e 4° dito; il 1° dito presenta più o meno malformazioni. Oltre alle sindattilie, sono evidenti varie lesioni ossee come la fusione delle falangi distali che inoltre appaiano alterate; le falangi del pollice sono fuse e formano un unico osso quadrangolare. Spesso troviamo la fusione delle falangi in un unico osso anche nel 1° dito del piede, mentre le falangi delle altre dita mostrano le stesse anomalie di quelle della mano.
Può esserci un moderato accorciamento degli arti superiori e limitazione della funzione delle grosse articolazioni; a volte si presentano associate malformazioni cardiache, intestinali, o renali. Molti pazienti hanno notevole ritardo mentale. La maggioranza dei casi sono sporadici, ma sono stati osservati anche rari casi familiari (Smith et al., 1982; Stricker et al., 1990; Slaney et al., 1996; Filkins et al., 1997).
La sindrome di Pfeiffer è caratterizzata da turribrachicefalia, ipertelorismo, proptosi e ipoplasia mediofacciale. Nei casi più gravi, la craniostenosi complessa porta trifogliocefalia, proptosi pronunciata e coinvolgimento del sistema nervoso centrale. Tale forma più severa è stata definita Pfeiffer di tipo II (Cohen, 1993).Il termine trifogliocefalia è stato coniato per descrivere la morfologia anomala del cranio che assume la forma di trifoglio, a causa della simultanea sinostosi delle suture coronale e lambdoidea (David et al, 1982). La trifogliocefalia è stata osservata sia come caso isolato sia come espressione delle forme sindromiche. Generalmente, la presenza di ampi pollici e marcata deviazione di corti alluci, di brachidattilia e di una parziale sindattilia cutanea delle mani e/o dei piedi permettono di distinguere la sindrome di Pfeiffer da altre condizioni craniosinostotiche (Smith et al., 1982; Introzzi, 1984; Stricker et al., 1990; Muenke et al., 1994).
La sindrome di Jackson-Weiss è caratterizzata da craniosinostosi in combinazione ad un ampiospettro di anomalie eterogenee delle dita dei piedi quali: il 1° metatarso ampio e corto, falangi degli alluci ampie, anomalie nella forma della ossa tarsali e fusione delle ossa metatarsali e/o tarsali. I pazienti affetti presentano un dismorfismo facciale con uno spostamento asimmetrico verso il basso e una severa brachicefalia. Normalmente sono assenti anomalie nelle dita delle mani (Smith et al., 1982; Introzzi, 1984; Stricker et al., 1990; Cohen, 1995).
La sindrome di Saethre-Chotzen è caratterizzata da una asimmetria facciale dovuta a plagiocefalia, in combinazione a brachidattilia e sindattilia cutanea delle mani (2° e 3° dito e/o 3° e 4°) e delle dita dei piedi. Possono essere presenti anche altre anomalie come un ritardo nello sviluppo mentale e malformazione di alcuni organi (rene, cuore) (Smith et al., 1982; Introzzi, 1984; Stricker et al., 1990; Reardon et al., 1994).
Fattori molecolari coinvolti
Sebbene diversi fattori, come ipertiroidismo, agenti teratogeni, malformazioni o disordini ematologici possono portare all’insorgenza della malattia, le condizioni monogeniche sono comunque quelle coinvolte più frequentemente nell’eziologia delle craniosinostosi. Fino ad ora sono stati identificati tre geni o famiglie di geni considerati essere la causa di alcune sindromi craniosinostotiche: i geni per la famiglia dei recettori di membrana per i fattori di crescita dei fibroblasti (FGFR1-3), e i geni per due fattori di trascrizione, MSX2 e TWIST.
Fattore MSX2
MSX2 è un fattore di trascrizione il cui gene è stato clonato per omologia con il gene msh di Drosophila. Esso contiene un omeodominio altamente conservato che è coinvolto sia nel legame al DNA che nell’interazione con proteine (Jabs et al., 1993).
Il gene MSX2 mappa nel cromosoma umano 5q34-35. Coerentemente con il suo coinvolgimento nella funzione e nella chiusura delle suture, l’mRNA di MSX2 è stato localizzato a livello delle suture e in altri siti durante lo sviluppo craniofacciale del topo. La struttura di MSX2 contiene un omeodominio centrale che si pensa sia coinvolto nell’interazione con specifici siti nelle regioni del promotore dei geni bersaglio (Davidson et al., 1995).
Una sindrome craniosinostotica, denominata tipo Boston, a trasmissione autosomica dominante, è stata associata ad una mutazione di MSX2 di tipo missenso che risulta nella sostituzione Pro148His all’interno dell’omeodominio in posizione 7 (Jabs et al, 1993;Muragoki et 1998). Recentemente è stato dimostrato che la proteina nella quale è presente questa sostituzione aminoacidica mostra differenze nelle proprietà di legame al DNA senza però alterare la specificità di legame alla sequenza bersaglio: questa mutazione infatti sembra influire sulla proteina riducendo la sua velocità di dissociazione dal DNA (Golden et al, 1996). Questi ed altri risultati hanno portato all’ipotesi che lo sviluppo craniofacciale sia sensibile al dosaggio della proteina e che la maggiore affinità di legame al DNA della proteina con la mutazione Pro148His possa mimare l’effetto di un aumento di dosaggio sufficiente a causare craniosinostosi ma non alterazioni più gravi.
TWIST
Mutazioni nel gene TWIST sono state recentemente associate alla sindrome di Saethre-Chotzen. TWIST è l’omologo del gene twist di Drosophila, ed è membro di una vasta famiglia di fattori di trascrizione bHlH, cioè caratterizzate dalla presenza di un dominio di residui aminoacidici basici seguito da tre domini caratteristici “elica–ansa-elica”(Baylies MK et al., 1996).
Il locus per questa sindrome è stato mappato nel cromosoma 7p21-p22; il gene TWIST, che codifica per un un fattore di trascrizione con dominio bHLH, è stato mappato nel cromosoma umano 7p21 ed è stato perciò ritenuto un importante candidato per l’insorgenza di questa sindrome. L’analisi di questo gene, in pazienti affetti da tale patologia, ha infatti individuato mutazioni non senso, missenso, delezioni e inserzioni. Queste mutazioni risiedono prevalentemente in quelle porzioni del gene che codificano per la regione basica di legame al DNA, e i domini HLH. L’osservazione che topi eterozigoti twist+/- mostrano caratteristiche fenotipiche analoghe a quelle presenti in pazienti affetti dalla sindrome di Saethre-Chotzen con una delezione nella regione 7p, ha portato a formulare l’ipotesi che il meccanismo molecolare delle mutazioni nel gene TWIST possa essere la perdita di funzione di un allele (aploinsufficienza). Dall’analisi di sequenza è stata individuata una vasta gamma di mutazioni in pazienti con sindrome di Saethre-Chotzen. Il gene TWIST contiene due esoni ed un introne lunghi rispettivamente 772bp, 645bp e 538bp. Tutte le mutazioni puntiformi sono localizzate dentro la regione codificante; non sono state identificate mutazioni nei siti di splicing, nell’introne o nel secondo esone (El Ghouzzi et al, 1997). Le mutazioni non senso sono state riscontrate prevalentemente nell’estremità 5’ della sequenza codificante; esse precludono la traduzione dei domini di legame al DNA e elica-ansa-elica. La presensa di una mutazione missenso ed una inserzione nel quadro di lettura di 21 coppie di basi al 5’ dei domini di legame al DNA suggerirebbero che questa regione, sebbene al di fuori dei domini funzionali già caratterizzati, sia comunque importante per la struttura e la funzione della proteina. In modo simile, la presenza di una mutazione no senso al 3’ nel dominio della seconda elica al codone 181 suggerisce che anche il segmento carbossiterminale sia necessario per la corretta funzionalità della proteina (Howard et al, 1997). Dall’analisi di ibridizzazione in situ con sonda fluorescente (FISH) e di southern blot sono state identificate grosse delezioni con completa perdita della sequenza codificante (Rose, 1997). Polimorfismi apparentemente non associati alla patologia nel gene TWIST sono stati identificati attraverso l’analisi di 160 alleli da soggetti non imparentati ma affetti da craniosinostosi. Questi polimorfismi includono una mutazione missenso conservata, un cambiamento di nucleotide in una posizione oscillante, e due differenti inserzioni di tre coppie di basi che portano all’aggiunta di un residuo di glicina ad un tratto di 5 glicine. Un’inserzione di 21 coppie di basi dentro una di queste regioni ricche di glicina, risulta nell’aggiunta di 5 residui di glicinae di una alanina. Tutti i polimorfismi si trovano nel 5’ dei domini funzionali, e non sono stati notati polimorfismi all’interno dell’estremità 3’ dei domini di legame al DNA e dei domini elica-ansa-elica (Kasparcova et al 1998; Gripp et al 2000).
Recettori dei fattori di crescita dei fibroblasti (FGFR)
I fattori di crescita dei fibroblasti (FGF) costituiscono una famiglia di proteine, strutturalmente correlate, implicate in molteplici processi biologici quali l’angiogenesi, lo sviluppo embrionale e la trasformazione tumorale (Mason, 1994; Muenke et al., 1995). La loro azione, che può essere diretta a diversi tipi cellulari di origine mesodermica e neuroectodermica, stimola le cellule bersaglio alla proliferazione, all’inibizione o all’induzione del differenziamento, e alla migrazione cellulare (Muenke et al., 1995; De Moerlooze et al., 1997). Gli FGF interagiscono con proteoglicani eparan-solfati nella matrice extracellulare che hanno un ruolo importante nel modulare il legame dei fattori di crescita con i loro recettori ad alta affinità: i recettori per i fattori di crescita dei fibroblasti (FGFR), che mediano la trasduzione del segnale intracellulare. La famiglia degli FGFR è costituita da quattro recettori transmembrana, FGFR1, FGFR2, FGFR3 e FGFR4, codificati da 4 distinti geni localizzati in diversi cromosomi (rispettivamente 8, 10, 4, e 5); essi mostrano un elevato grado di conservazione sia a livello di struttura genica che a livello di sequenza aminoacidica. Nonostante la relativamente alta identità di omologia nella loro sequenza aminoacidica, gli FGFR differiscono per la loro affinità di legame ai diversi FGF. Questa diversa specificità per i ligandi e la fine regolazione della loro espressione nei diversi tessuti spiega il molteplice ruolo del segnale degli FGF nel differenziamento e nell’organogenesi (Fernig et al., 1994; Muenke et al., 1995).
Gli FGFR hanno una struttura comune che consiste in una porzione extracellulare contenente un segnale peptidico, tre domini immunoglobulino-simili (Ig simile I-III) ciascuno dei quali possiede una coppia di residui di cisteina altamente conservati nei diversi recettori, e una sequenza di residui aminoacidici acidi tra il primo e il secondo Ig simile (acidic box); una regione transmembrana; una regione intracellulare che consiste in un dominio ad attività tirosina-chinasica diviso da un inserto chinasico e domini di interazione con proteine di regolazione citoplasmatiche. Il meccanismo coinvolto nella trasduzione del segnale ancora oggi non è del tutto conosciuto. Il primo passo della trasduzione del segnale consiste in una omo- o eterodimerizzazione degli FGFR indotta dal legame del FGF. A questa dimerizzazione fanno seguito eventi di auto- e transfosforilazione che si verificano nel dominio chinasico. La presenza dell’eparan solfato nella matrice extracellulare aumenta l’affinità degli FGF per i recettori: questo proteoglicano infatti induce cambiamenti conformazionali sia nel nel ligando che nel recettore, favorendo il loro riconoscimento reciproco. I recettori una volta attivati possono fosforilare proteine di regolazione che attivano cascate di segnali in vie di trasduzione ramificate quali quelle associate alla proteina Ras o alla fosfolipasi C (Mason, 1994; Heldin, 1995).
Ciascun membro della famiglia degli FGFR è caratterizzato da un gran numero di isoforme che originano principalmente attraverso interventi di “splicing” alternativo del trascritto. Diverse regioni del messaggero sono soggette a questa classe di processamento, in particolare sembra avere un ruolo fondamentale nella funzione recettoriale, la scelta della sequenza che andrà a codificare per la seconda metà del terzo dominio immunoglobulino-simile. Il terzo dominio Ig simile è codificato per la sua metà amminoterminale da un esone invariante denominato IIIa, il quale può essere unito per un evento di “splicing” alternativo ad uno di due esoni , adiacenti localizzati a valle, indicati IIIb e IIIc, entrambi codificanti per la porzione carbossiterminale del dominio. La specificità di legame del recettore per il ligando è determinato proprio dalla scelta tra i due esoni al 3’ sottoposti a “splicing”. Alcuni FGF legano specificatamente singole isoforme recettoriali IIIb o IIIc (come ad esempio FGF3, FGF5 e FGF7), altri invece sono in grado di legare entrambe le isoforme del recettore (come FGF1). Comunque la maggior parte degli FGF lega e attiva almeno due o tre recettori, normalmente gli isotipi IIIc o IIIb (Wilkie et al., 1995).
Isoforme FGFR
Attraverso “splicing” alternativo vengono prodotte almeno 6 isoforme del FGFR1; le varianti possono essere prodotte mediante delezioni di soli due aminoacidi di tutto il dominio immunoglobulinico, della sequenza segnale e dell’intera porzione intracellulare, della regione transmembrana e di parte del terzo dominio Ig simile. Quest’ultima variante non è più incorporata nella membrana, ma viene secreta dalla cellula. Un’altra isoforma del FGFR1 presenta una troncatura nel dominio tirosin chinasico (Fernig et al., 1994).
Anche le isoforme del FGFR2 sono generate da simili processi di “splicing” alternativo. L’isoforma IIIb dell’FGFR2 (e in generale degli FGFR) viene espressa nelle cellule epiteliali, l’isoforma IIIc è invece espressa nelle cellule di origine mesenchimale. In maniera reciproca, i ligandi per le isoforme IIIb dei recettori sono generalmente espressi nel mesenchima, mentre i ligandi per le isoforme IIIc dei recettori vengono espresse nell’epitelio (Goldfarb, 1996). Questo suggerisce che gli FGF siano in grado di mediare una comunicazione bidirezionale tra il mesenchima e l’epitelio.
Le mutazioni nei geni FGFR1-3 responsabili per forme non sindromiche e per le sindromi di Apert, Crouzon, Pfeiffer e Jackson-Weiss forniscono la prima evidenza genetica che il segnale del FGF possa svolgere un ruolo importante nella morfogenesi degli arti e in quella craniofacciale.
Coinvolgimento degli FGFR nello sviluppo craniofacciale e degli arti
Esistono consistenti evidenze sperimentali che hanno dimostrato un ruolo fondamentale per gli FGF nello sviluppo degli arti. L’applicazione ectopica del FGF1, FGF2 o FGF4 nel mesoderma della piastra laterale nel fianco di embrioni di pollo induce la formazione completa degli arti in quella sede (Cohn et al., 1995).
Durante lo sviluppo dello scheletro nel topo sono espressi sia FGFR1 che FGFR2. FGFR1 è espresso lungo il mesenchima dell’arto, nella zona di alta proliferazione delle cellule mesenchimali. L’FGFR1 e l’FGFR2 vengono coespressi nelle strutture preossee e precartilaginee, come per esempio durante la formazione delle ossa craniofacciali. L’FGFR3 invece viene espresso durante l’ossificazione endocondrale delle ossa lunghe. Durante lo sviluppo degli arti l’FGFR1 viene espresso in tutto il mesenchima mentre invece l’espressione del FGFR2 è confinata allo strato ectodermico (Goldfarb M, 1996).
Una serie di indagini sulle basi genetiche delle craniosinostosi e di altri disordini che interessano lo scheletro hanno fornito indicazioni significative per la comprensione del ruolo degli FGFR nello sviluppo sia degli arti che del complesso craniofacciale (Yamaguchi et al., 1995). La loro posizione cromosomica e la loro espressione spazio-temporale durante lo sviluppo embrionale ha reso i geni per gli FGFR possibili ottimi candidati per tali disordini dello sviluppo (Winter, 1995, Yamaguchi et al., 1995 ).
Nella regione del cromosoma 4p16, che contiene il gene FGFR3, studi genetici di associazione hanno permesso di localizzare i loci di tre osteocondroplasie: l’acondroplasia, che è la forma più comune di nanismo, l’ipocondroplasia e la displasia tanatoforica. L’analisi della sequenza del FGFR3 in pazienti acondroplasici ha portato all’identificazione di una mutazione nella regione transmembrana del gene che probabilmente altera la struttura idrofobica ad a-elica e quindi la fusione della dimerizzazione del recettore in seguito al legame con il ligando. Analisi di associazione hanno permesso di indicare il gene FGFR2 come possibile gene candidato per le sindromi craniosinostotiche di Apert, Crouzon (CS) e Jackson-Weiss (JWS), che mappano sul cromosoma 10q25-q26. La sindrome di Pfeiffer (PS), è stato dimostrato essere geneticamente eterogenea perché i loci ad essa associati mappano in posizione 10q25-q26 (FGFR2), e in 8cen (FGFR1), facendo supporre il coinvolgimento di entrambi i geni.
Ad alcuni pazienti affetti da sindrome di Pfeiffer, è stata associata una singola mutazione missenso in FGFR1 nella sequenza che codifica per la porzione extracellulare tra il II e il III dominio Ig-simile. In pazienti Pfeiffer privi della mutazione in FGFR1 sono state identificate mutazioni nel gene FGFR2. Consistentemente con il fenotipo Pfeiffer, entrambi i geni vengono coespressi nei tessuti colpiti da questa sindrome come gli arti e le regioni craniofacciali . Tutte le mutazioni identificate negli FGFR agiscono in modo dominante ed la maggioranza comportano la sostituzione di un residuo aminoacidico nella porzione extracellulare del terzo dominio Ig simile (IgIII) dei recettori FGFR1-3.
Al gene FGFR4, fino ad oggi, non sono stati ancora associati disordini dello scheletro

genetica molecolare