
Sindrome di Alport
Nefrite ereditaria scoperta da Cecil Alport nel 1927 quando presentava a Londra lo studio di una famiglia i cui soggetti di sesso maschile avevano ematuria persistente e sordità di tipo percettivo.
Manifestazioni Renali:
1.Ematuria
2.Proteinuria
3.Ipertensione
4.Insufficienza renale cronica
Manifestazioni Extra-renali:
1.Deficit uditivi
2.Difetti oculari
Patogenesi:
Alterazione della composizione del collagene IV,
uno dei principali componenti delle membrane basali
Il Collagene Costituente del tessuto connettivo, insieme a elastina, fibrillina e proteoglicani, tutti prodotti da fibroblasti, condroblasti, osteoblasti La proteina + abbondante nei mammiferi (25% del loro peso): presente in cartilagine, tendini, legamenti, osso, matrice extracellulare, membrane basali, cornea e cristallino. Filogeneticamente molto antico: già presente nei Poriferi (600 milioni di anni fa)
Struttura del collagene
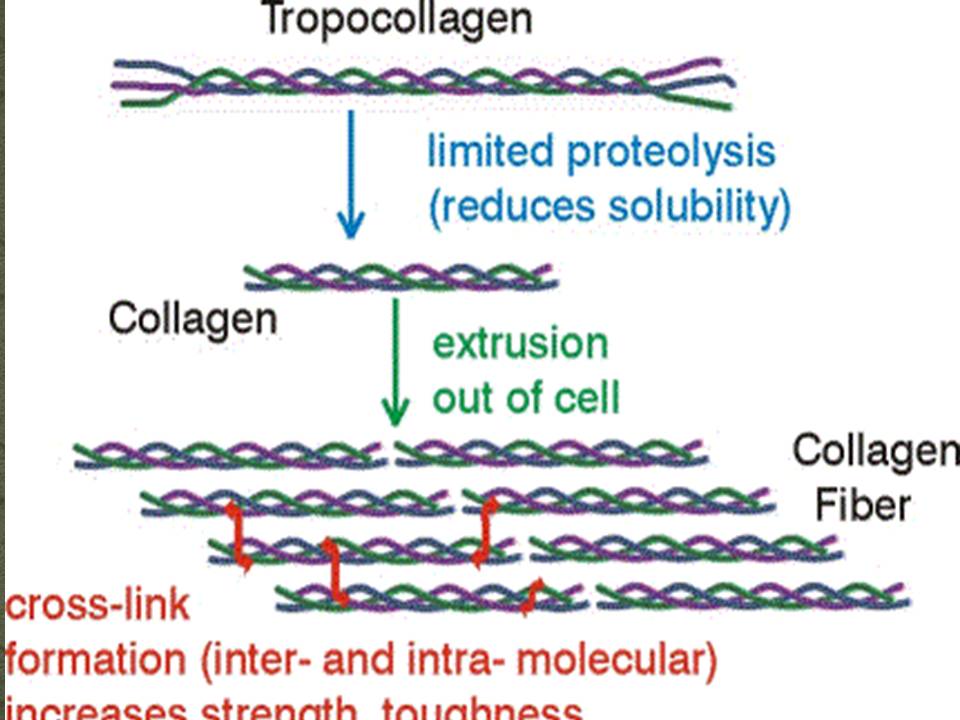
Unità costitutiva è il tropocollageno (~ 300 kDa):
formato dall’unione di 3 catene polipeptidiche avvolte in una tripla elica destrorsa molto stretta (elica del collagene)
molto resistente alla trazione, ma non elastica (cime delle navi).
La polimerizzazione spontanea (extracellulare) delle unità di tropocollageno forma le fibrille collagene.
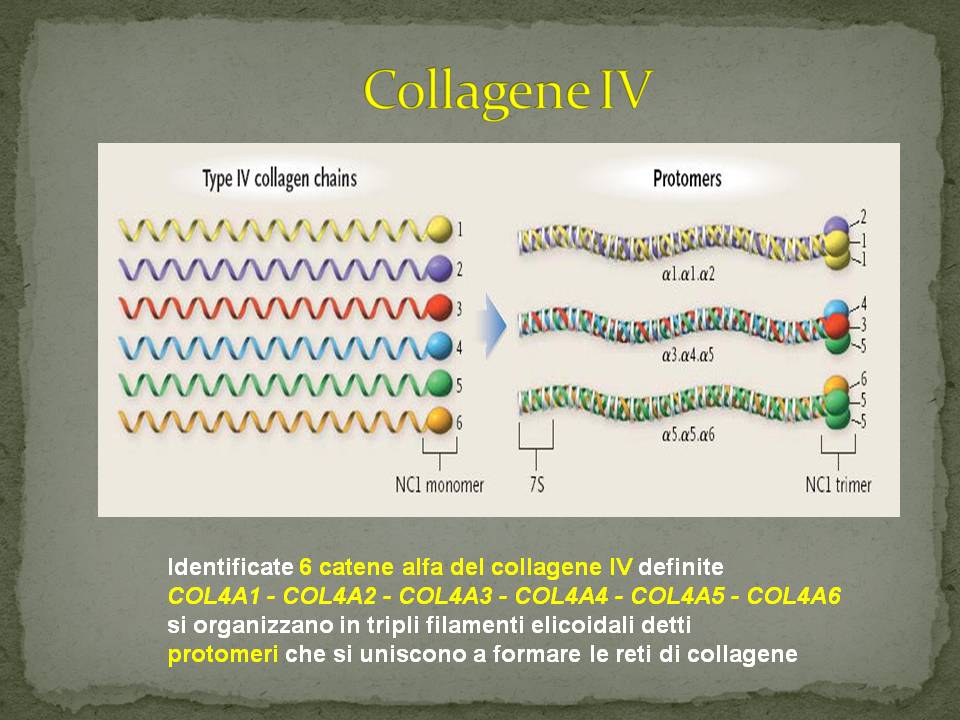
Tipi di collageno (almeno 28 tipi)
I protomeri si uniscono a formare tre tipi di network-collagene:
1) network 1.1.2/1.1.2 presente in tutte le membrane basali
2) network 3.4.5/3.4.5 nelle membrane basali glomerulari,
in alcune membrane basali tubulari,
coclea, occhio, polmone, testicolo
3) network 1.1.2/5.5.6 nella pelle, nel muscolo liscio e
nella capsula di Bowman
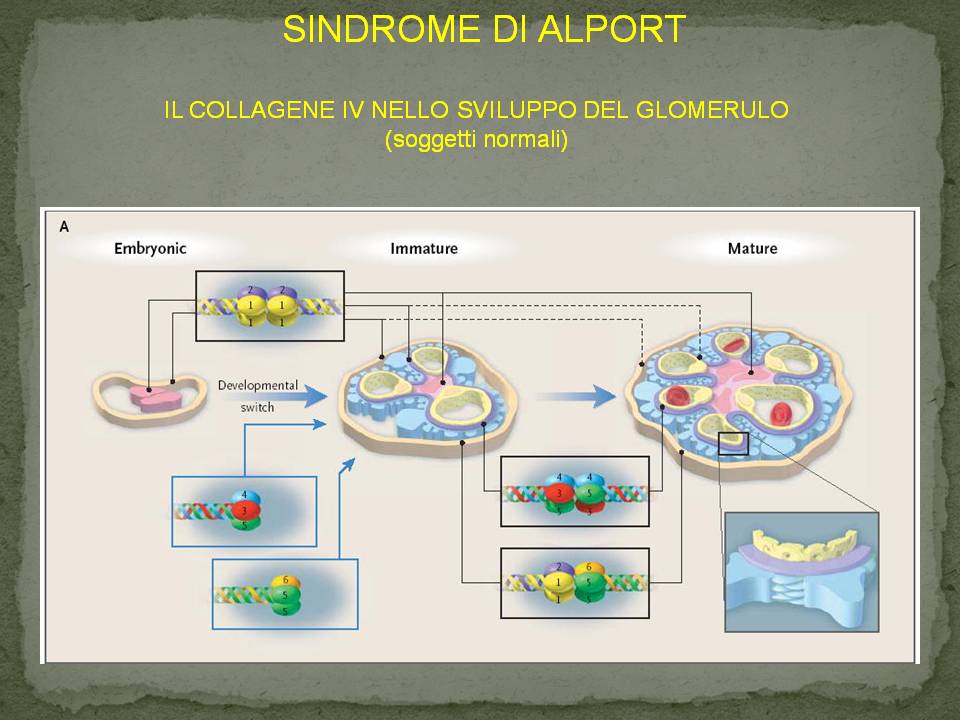
La lamina densa della membrana basale glomerulare è costituita da un intreccio delle catene aminoacidiche a3, a4 e a5.
La presenza di una mutazione di una delle 3 catene
comporta un’ alterazione della stabilità degli eterotrimeri del collagene IV, con conseguente sua alterazione anatomica e morfologica.

Mutazioni identificate
- Mutazioni missenso
Modificano il significato di un codone e si ha un aminoacido al posto di un altro nella proteina codificata
- Mutazioni di non senso o di stop
Si realizza un codone di stop (UAA,UAG,UGA) con conseguente proteina tronca
- Mutazioni frameshift
Si realizza uno sfasamento della lettura a causa di inserzioni o delezioni
- Mutazioni che alterano lo splicing
Alterazione dell’mRNA e di conseguenza della proteina codificata
SINDROME DI ALPORT (nefrite ereditaria)
- prevalenza della mutazione genetica 1/5.000 – 1/10.000
- 1-2% dei casi di IRC terminale
- ereditarietà X-linked nell’85% dei casi
- autosomico recessivo nel 10% dei casi
- autosomico dominante nel 5% dei casi
Alport X-linked è responsabile dell’ 85% dei casi.
Manifestazioni cliniche nella sindrome Alport X-linked
Più di 300 mutazioni del COL4A5 sono state finora identificate.
Le mutazioni possono dare origine a proteine assenti o troncate
I geni mutanti X-linked sono completamente espressi nei maschi che hanno un solo cromosoma X ( emizigoti)
A causa dell’inattivazione casuale della X (lyonizzazione), il carattere è variabilmente espresso nelle donne eterozigoti
Il carattere non è mai trasmesso dal padre al figlio maschio
Le figlie di un uomo affetto sono portatrici obbligate
I figli maschi di una donna portatrice hanno il 50% di probabilità di essere affetti
Le figlie femmine di una donna portatrice hanno il 50% di probabilità di essere portatrici.
Correlazione genotipo fenotipo?
Recente studio dell’ “European Community Alport Syndrome Concerted Action”: studiati 401 pz di sesso maschile appartenenti a 195 famiglie con mutazioni sul gene COL4A5:
Pz con mutazioni “missenso”: prognosi migliore (50-70% dei pz andranno in IRC terminale + defictit uditivi prima dei 30 anni)
Pz con delezioni, mutazioni non senso o con cambiamento del “reading frame”: prognosi peggiore (90% dei pz andranno in IRC terminale + defictit uditivi prima dei 30 anni
S. di Alport X-LINKED
(aspetti clinici)
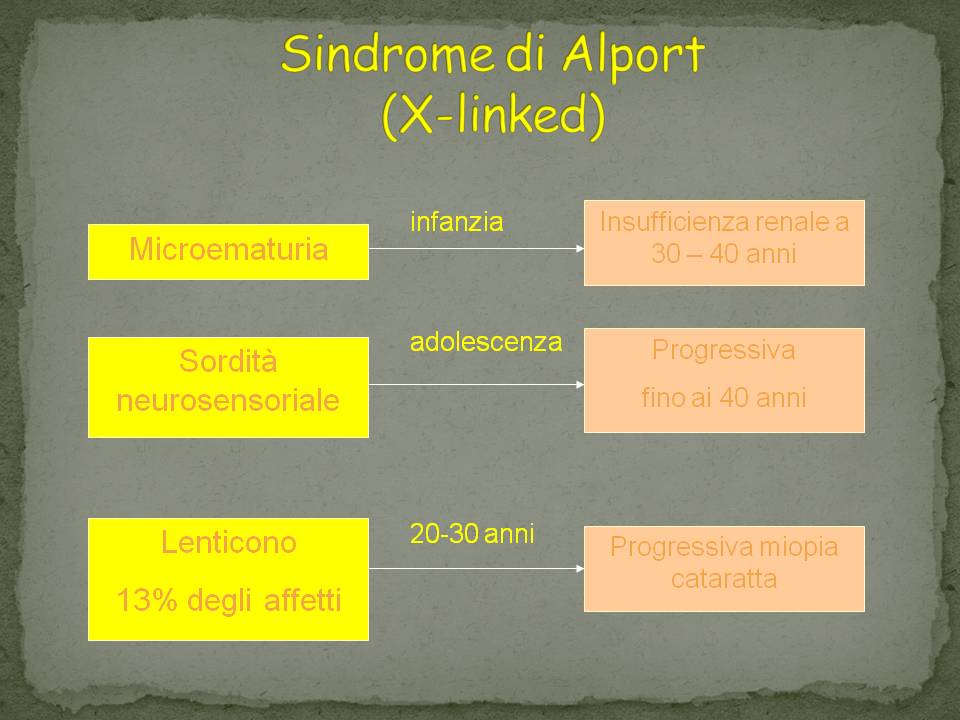
La progressione del danno renale è più grave nei pazienti maschi, che presentano ematuria nella prima infanzia, molto spesso sviluppano una sordità neurosensoriale progressiva durante l’età scolastica e, di solito, un’insufficienza renale allo stadio terminale nei primi vent’anni, con o senza anomalie oculari.
Il quadro clinico potrà essere modesto, lieve o assente nelle femmine e solo una minima parte di loro sono affette in modo grave, a seconda che prevalga il cromosoma X “sano” oppure quello “patologico” secondo la legge di Lyon
Le femmine saranno comunque portatrici.
SINDROME DI ALPORT (nefrite ereditaria)
- prevalenza della mutazione genetica 1/5.000 – 1/10.000
- 1-2% dei casi di IRC terminale
- ereditarietà X-linked nell’85% dei casi
- autosomico recessivo nel 10% dei casi
- autosomico dominante nel 5% dei casi

Sindrome di Alport AR (aspetti clinici)
sia i maschi che le femmine possono essere colpiti con uguale probabilità
tutti i soggetti omozigoti presentano microematuria con possibili episodi di macroematuria e possibile proteinuria
i soggetti eterozigoti sono asintomatici o possono avere microematuria
la perdita della funzionalità renale è precoce, indipendentemente dal sesso
SINDROME DI ALPORT (nefrite ereditaria)
- prevalenza della mutazione genetica 1/5.000 – 1/10.000
- 1-2% dei casi di IRC terminale
- ereditarietà X-linked nell’85% dei casi
- autosomico recessivo nel 10% dei casi
- autosomico dominante nel 5% dei casi

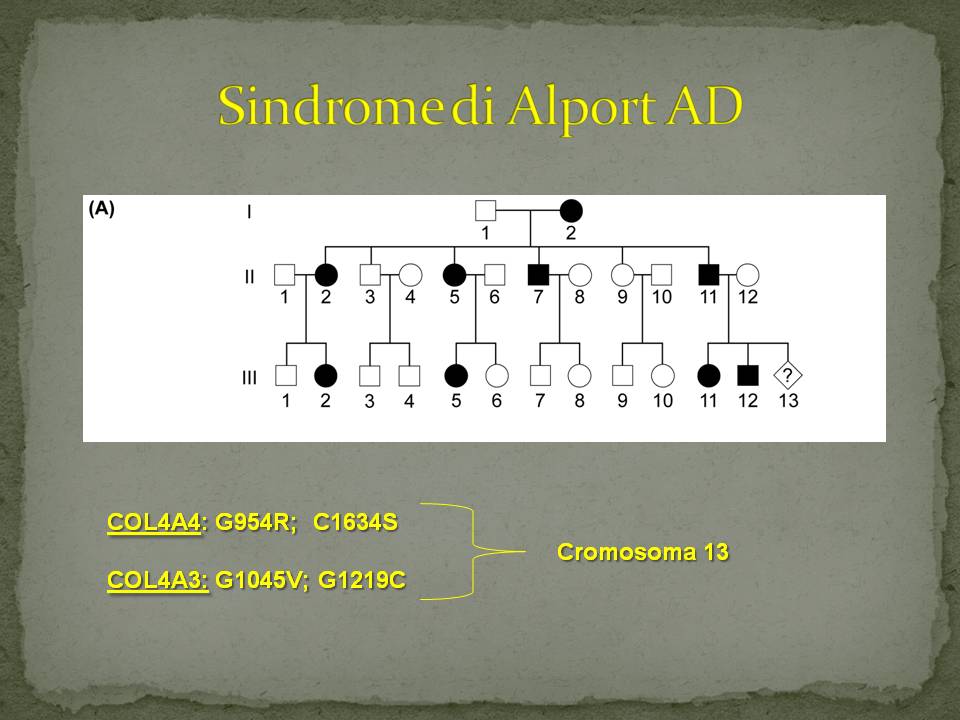
Sindrome di Alport Autosomica Dominante (aspetti clinici)
- Gli individui affetti possono avere genitori affetti
- Nella stessa famiglia possono essere affetti indifferentemente sia maschi che femmine
- Da un genitore affetto eterozigote possono nascere teoricamente il 50% di figli affetti e il 50% di figli non affetti
- Tipicamente si possono osservare individui affetti in tutte le generazioni
Caratteristiche Mutazioni Autosomiche Dominanti
COL4A4: con sostituzione di una Glicina (anfotera)nel dominio collagenico della proteina con una Arginina (idrofila)
COL4A4: con sostituzione di una Cisteina (idrofila) nella regione C-terminale non-collagenica della proteina con unaSerina
COL4A3: con sostituzione di una Glicina nel dominio collagenico della proteina con una Valina (idrofoba)
COL4A3: con sostituzione di una Glicina nel dominio collagenico della proteina con una Cisteina
SINDROME DI ALPORT
Diagnosi
- Va sospettata nei p. con ematuria (con o senza deficit uditivo)
e anamnesi familiare positiva
- Per la conferma necessaria biopsia renale
- L’assenza contemporanea della catene alfa3 alfa4 alfa5
è altamente specifica per la s. di Alport
- L’assenza della catena alfa5 nella biopsia cutanea
è altamente specifica per la forma X-linked
Poiché nella MB epidermica sono normalmente espresse
le catene alfa5 alfa6, ma non le catene alfa3 alfa4,
con la biopsia cutanea può essere distinta la forma X-linked
da quelle autosomiche
- diagnosi molecolare: nei p. con mutazione genetica
caratterizzata nei familiari
Terapie
Trapianto renale
Dialisi
Terapia di sostegno
Sperimentazioni di cellule staminali del midollo spinale